
Introducing PLUG: A Compiler for Leakage-free Protein Function Benchmarking
How is protein function predicted?
Accurately predicting protein function remains one of the central challenges in biological machine learning (ML). Despite recent successes of protein language models (pLMs) and structure predictors, one question remains unclear: which aspects of a protein’s function do current models capture and how well?
Protein function is intrinsically multifaceted; a single protein can perform multiple functions, including catalysis, ligand binding, and allosteric signaling, which rely on environmental contexts such as expression, pH, macromolecular crowding, and other perturbations. Descriptors of protein function range from coarse-grained labels like Gene Ontology (GO) terms and Enzyme Commission identifiers to specific properties such as catalytic activity, binding affinity, and specificity. Experimental assays typically measure only one of these properties at a time, meaning no single measurement can capture the full functional landscape.
A well-rounded protein function model should predict general function (e.g. GO terms), but also more specific properties such as allostery, binding affinity, and catalytic activity. However, most models are developed and evaluated against a single downstream task, and strong performance on one benchmark dataset does not always translate to success on another. Protein foundation models aim to learn broad representations of proteins, but often struggle to outperform narrower models on functional prediction, making their generalization difficult to assess. Towards better generalized modeling of protein function, it is necessary to build valid tests that evaluate model performance as a function of task specificity and training similarity.
The problem of data leakage
Defining data leakage for protein function prediction is itself a challenging problem. Leakage is often treated as a simple sequence similarity problem, where proteins above a chosen sequence identity threshold are removed from the training set. In practice, however, leakage can occur in nearly countless ways. Proteins with low overall sequence identity may still share highly conserved domains, catalytic sites, or other motifs that are hard to detect. Conversely, proteins with divergent sequences can adopt very similar structures, allowing a structural model to associate a particular topology with a functional label, thereby missing the underlying mechanism. In mutational datasets, holding out individual variants while training on neighboring mutations or different substitutions at the same residue often measures interpolation within a mutational landscape rather than true generalization. Even seemingly straightforward notions of sequence leakage become complicated by multidomain proteins, proteins with large and highly conserved non-active site regions, and experimentally dictated expressions of the same mechanism (i.e. an scFv vs. a Fab), making it difficult to define what constitutes an independent training example.
Benchmark quality also degrades over time. As new experimental datasets are released, proteins that once only appeared in test sets are gradually added to new training corpora. Without careful filtering, models may inadvertently train on proteins that are highly similar or even identical to benchmark examples, inflating reported performance. Maintaining meaningful evaluation therefore requires continually rebuilding training sets as new data become available rather than relying on static benchmark splits.
PLUG: Protein Leakage-free evaluation for Unbiased Generators of function
To better discern a model’s functional knowledge, we developed PLUG (Protein Leakage-free evaluation for Unbiased Generators of function), an intentionally modular framework for constructing leakage-controlled training sets across continuously evolving collections of functional benchmarks. Rather than introducing a single new benchmark, PLUG provides a reproducible framework for compiling many.
PLUG has two primary components. First, it aggregates multifaceted validation datasets spanning biologically relevant functional axes into a common space with standard ML operational pipelines. Second, it constructs leakage-free training subsets from either a sequence reservoir (UniRef90), a structure reservoir (PDB), or both, while explicitly checking for overlap with one, multiple, or all functional test sets.
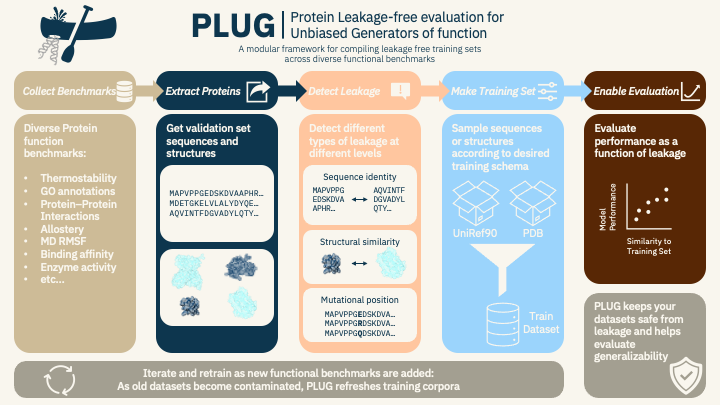
This produces a set of sequences and structures for model training that excludes proteins similar to the evaluation sets, ensuring safe benchmarking across different protein functions. PLUG easily accepts new benchmarks, requiring only an adapter for new data, allowing training sets to be rebuilt as the benchmark suite expands. This enables the community to maintain continually evolving evaluation targets that resist gradual contamination.
Functional benchmark suite
The initial suite of benchmarks spans multiple views of protein function across scales and domains. Examples currently supported include thermostability through the megascale dataset, deep mutational scan fitness from ProteinGym, GO term annotation from CAFA, allosteric site annotation from Allobench and PASSerRank, protein–protein interactions (PPI) from STRING and others, binding affinity from PDBBind and BindingDB, and RMSF from ATLAS.
Together, these tasks evaluate different aspects of protein function. They include interactions between residues within the same protein domain, such as in Allobench, interactions between proteins, as in STRING, and protein-level properties such as expression and enzyme classification. These test sets also vary in label noise, experimental conditions, and downstream applications, providing a broader basis for evaluating protein representation learning than optimizing for a single benchmark metric.
Leakage-controlled training set construction
Evaluating data leakage is inherently difficult because fully unbiased sampling is nearly impossible. For example, the PDB itself reflects decades of structural biology focused on experimentally tractable proteins. Nevertheless, stringent screening criteria can produce high-fidelity training sets for assessing generalization.
The core technical contribution of PLUG is a leakage filtering pipeline operating directly on a reservoir of sequences, structures, or both (e.g. UniRef90 and PDB). PLUG is applied to this reservoir and any observation sufficiently similar to a protein in the evaluation set is removed (with default thresholds of MMSeqs2 ID > 0.2 or Foldseek TM-score > 0.5, both of which are user configurable), resulting in a leakage-controlled training set and rigorous evaluation set. Traditional methods rely solely on a unidirectional sequence similarity score or temporal holdouts to remove homologs from the training set, whereas PLUG checks for bidirectional leakage with tunable coverage across both sequence and structure.
Rather than exhaustively searching every entry in the reservoir, PLUG provides functionality to iteratively sample candidate training proteins that satisfy the desired training distribution. For example, training proteins can be sampled by sequence length, protein family, deposition date, or other user-defined criteria. Candidate proteins are repeatedly sampled and screened until the desired training set quota is reached.
Training sets are evaluated for contamination at the level of full proteins, domains, and subdomains using multiple coverage modes. In the case of a model trained on both sequence and structure, a union graph is built over sequence and structure, and entire clusters are removed until the training set is adequately safe. Thresholds and parameters are deliberately user-defined to characterize generalizability as a function of data leakage.
Looking ahead
PLUG is an open-source modular framework intended for continual development. Beyond sequence and structural homology filtering, future extensions include additional sampling strategies, improved ligand encoding, and expanded benchmark coverage aimed at conformational dynamics. In a forthcoming publication, we will describe the complete methodology, present the full framework, and evaluate current models across the benchmark suite.
We hope PLUG provides the community with a reproducible foundation for constructing leakage-controlled protein function benchmarks and contributes to more rigorous evaluation of protein function models.
Code:
Code is available on GitHub.